CRIMEtoYHU
Help
User Interface
By an HTML form, the user must enter the name of the human gene to analyze and
select one or more tumors from a list of primary sites from the COSMIC database.
For each type of cancer, the associated mutations will be extracted and analyzed.
Moreover, user can also add any other protein variants to be analyzed.
Each mutation must be inserted into the query form according to syntax notation defined by Human
Genome Variation Society (HGVS).
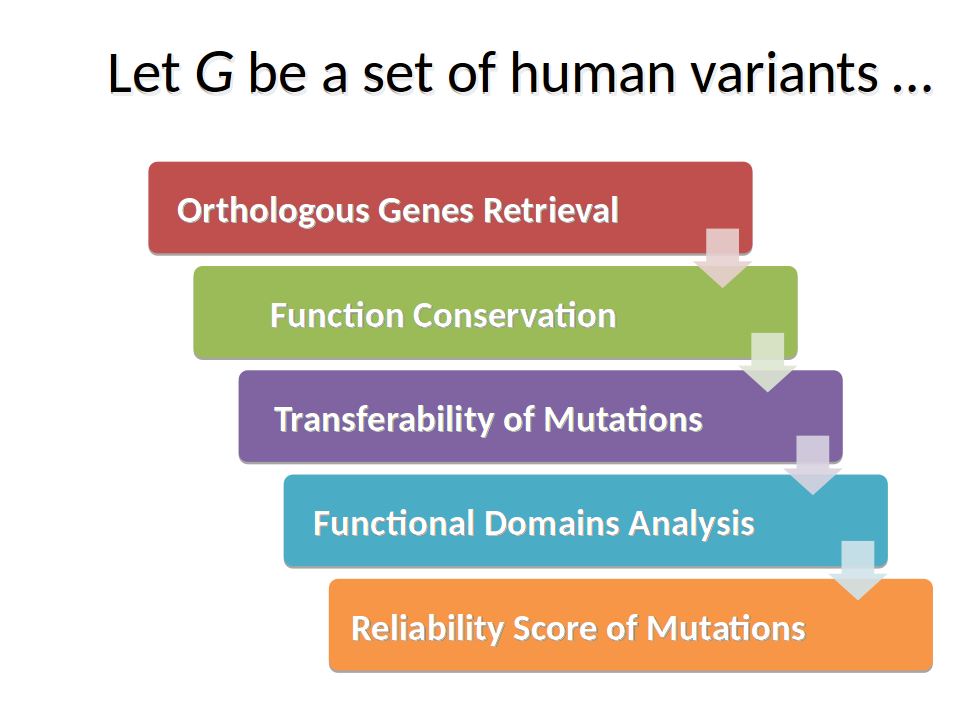
Step 1: Orthologous Genes Retrieval
The existence of an orthologue in yeast S.Cerevisiae of the human gene is the necessary condition for the process execution of mutations analysis. If no yeast homologous gene is found, the process is aborted.
Step 2: Function Conservation Analysis
The conservation of protein domains and functions is not a limiting condition to continue the analysis, but it is highly discriminating for the transferability of mutations in yeast homologous gene.
Functional conservation analysis compares the profiles of human and yeast homologous genes and then, reports the common functional domains conserved by the protein expressed by the orthologous gene.
The analysis examines the information on biological processes (BP), cellular components (CC) and molecular functions (MF) exploring Gene Ontology.
It also examines the annotated pathways in KEGG and Reactome databases.
Step 3: Analysis of Transferability of Mutations
From local instance of the COSMIC database, the tool extracts information of somatic mutations associated to one or more cancer types.
A key-point of the analysis concerns the assessment of the transferability of the human variants into yeast orthologous gene. The algorithm used by CRIMEtoYHU explores the areas of conservation by aligning the two amino acid sequence and localizes the position of human mutations.
In sequence alignment algorithms (both amino acids and nucleotides) a similarity value, dependent by the degree of conservation of the two sequences, is computed. The greater the amino acid conservation between yeast and human protein, the greater the numerical value of similarity.
By using the BLOSUM substitution matrices which assigns to each possible pair of amino acids a value that indicates their degree of similarity, we give information on the probability that an amino acid is substituted with another during evolution.
Step 4: Functional Domains Analysis
The sequence of amino acids from the yeast orthologue protein is analyzed through RESTful services running InterproScan of EBI.
The service identifies functional domains using 15 databases such as
Prodom, PRINTS, PIRSF, PfamA, SMART, TIGRFAM, PrositeProfiles, HAMAP, PrositePatterns, SuperFamily, SignalP, TMHMM, Panther, Phobius and coils.
The filtered occurrences of mutations, which enjoy the status of Transferability Mutation, will be located within the merged recognized domains.
The results will be displayed in both graphical and tabular format.
Step 5: Reliability Score of Mutations
Data obtained in the previous steps of the pipeline are processed to assign a weighted value of
Reliability Score to each variant, using an algorithm that computes the degree of conservation of
each mutation on the basis of the type of mutation, the Alignment Score, Conservation Score, Mutation
Score values and the number of recognized domains for each location of variants under consideration.
The lower the conservation of amino acids of the homologous protein is, the greater the numerical value of the Reliability Score.
Finally, the user can select the sequence of residues associated with the chosen humanized variant.
The results will be displayed in both graphical and tabular format.